神経心理学 - ページ 24

クラッベ病の症状、原因、治療
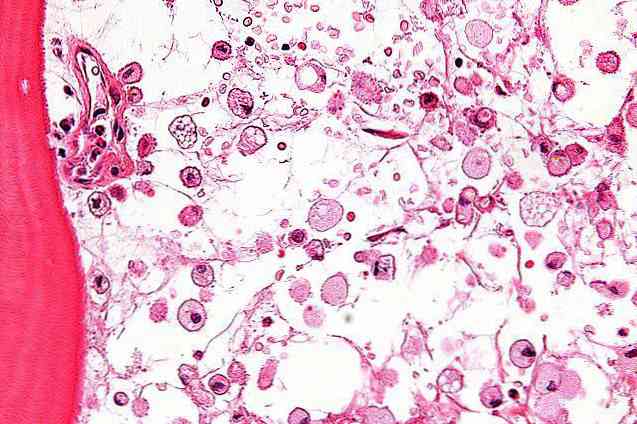
の クラッベ病 または球状白質ジストロフィーは、中枢神経系に影響を及ぼし、脳の白質またはミエリンの欠乏症を引き起こすまれな遺伝性疾患です。. それは非常に深刻で、しばしば致命的な病状であるという事実にもかかわらず、比較的未知の、遺伝的、遺伝的および退行性障害です。. このタイプの白質ジストロフィーは、神経系の髄鞘形成の欠如によって明らかにされ、赤字および他の神経学的障害の出現を引き起こします。.クラッベ病は男児と女児に等しく発症する。世界中で、この疾患の罹患率は10万人の出生につき約1人であると推定されている。しかし、スカンジナビア(50000人に1人)またはイスラエル(1000人に6人)のように、発生率がはるかに高い国があります。.クラッベ病の特徴白質ジストロフィー: "Leukós"、白+ "Dys"、不良または欠乏+ "Tréphein"から、栄養状態。白質の栄養障害グロボイド:グロボイド細胞との関係.クラッベ病としても知られています。 1916年にデンマークの神経内科医Knud Haraldsen Krabbe(1885-1965)からこの病状を有する患者の症例を報告した最初の人であることからその名前が付けられました。.クラッベ病は、白質ジストロフィーのグループの遺伝的疾患です。白質萎縮症は、ミエリンとしても知られている、脳の白質の産生または完全性に影響を及ぼす病状の一種です。.ミエリンは、神経細胞の軸索(電気的インパルスが駆動される場所)を裏打ちし、それらの周囲に鞘または層を形成し、それによって細胞の速度を向上させ、速める脳内の白い物質です。神経インパルスの伝達.ミエリンを生成する細胞エンベロープは、電気インパルスの正しい伝達を保証します。そのため、その完全性は中枢神経系および末梢神経系の機能に不可欠です。.通常の条件下では、ミエリンは軸索を覆って高抵抗の層を形成し、これは絶縁体として機能し、電気インパルスの正しい伝播を可能にする。もっとよく理解するには、ミエリンは電気ケーブルを覆うプラスチックコードのようなものです。. ミエリンの完全性が影響を受けると、細胞が脱髄されて神経インパルスの分散が起こり、その速度が遅くなるかまたはそれが起こらなくなると言われています。.ミエリンが一般的な形で危険にさらされたり悪化したりした場合、私たちは脱髄、または白質の欠如について話します。ミエリンは神経系におけるインパルスの正しい伝達を確実にするので、この状態の結果ははっきりと見えて劇的です。. このようにして、脱髄は知覚的、感覚的、認知的または運動的な障害を引き起こす可能性があります。多くの場合、完全麻痺や早期死亡を引き起こすために到着します。毎年、何千人もの人々が白血病のようなミエリンの完全性を危うくする障害に悩まされています.白質ジストロフィーが発生すると、ミエリンは中枢神経系の神経を適切に覆うことができず、したがって電気インパルスを十分に伝達することができません。.現在、科学界は、十二指腸疾患を白質ジストロフィーとして同定し、それらを5つの異なるグループに分類した:ペルオキシソーム性白質ジストロフィー、リソソーム性白質ジストロフィー、空洞性白質ジストロフィー、低ミエリン化白質ジストロフィーまたは未確定白質ジストロフィー。. そのタイプによる白質ジストロフィーの現在の分類は以下の通りです:ペルオキシソーム性白質ジストロフィー副腎白質ジストロフィー/副腎骨髄ニューロパチー.Refsum病(乳児または成人).ツェルヴェーガー症候群.新生児副腎白質ジストロフィー.リソソーム性白質ジストロフィー異染性白質ジストロフィー(またはLDM)舌状白質ジストロフィーまたはクラッベ病.空洞性白質ジストロフィーアレキサンダー病.カナバン病.CACH症候群.皮質下嚢胞を伴う巨頭脳白質ジストロフィー(MLC).低髄鞘形成性白質ジストロフィーPelizaeus-Merzbacher病.Pelizaeus-Merzbacher様疾患.痙性対麻痺2.低髄鞘形成および先天性白内障(またはHCC).未分類の白質ジストロフィーAicardi-Goutières症候群.未確定の白質ジストロフィー。原因遺伝子がまだ同定されていないか、同定の過程にあるもの.今日は、グロボイド型白質ジストロフィー、またはクラッベ病として知られるリソソーム型白質ジストロフィーの1つを説明し、知ることに焦点を当てます。.原因クラッベ病は、14番染色体(14q31)の小腕に位置するGALC遺伝子の変異によるものです。この遺伝子の突然変異を持っている人はガラクトセレブロシダーゼ、大量のミエリン脂質の異化作用に関与するリソソーム酵素と呼ばれる物質を十分に生産しません.ガラクトセレブロシダサの欠乏は、細胞傷害性物質であるサイコシンの蓄積を引き起こし、これがアポトーシス(プログラム細胞死)を引き起こす。非代謝脂質の蓄積は神経ミエリン保護鞘の成長に影響する. この物質(ガラクトセレブロシダーゼ)がないと、ミエリンは軸索の覆いを形成することができず、球状細胞群の形成が白質(中枢神経系および末梢神経系の両方)で起こり、神経結合が適切に機能しなくなります。.この病気の遺伝的な要素は劣性であり(それは遺伝子の2つのコピーを必要とします)、それは父から息子に伝染します。両方の親が不完全なGALC遺伝子突然変異を持っているならば、彼らの子供は突然変異したコピーを受け継がない25%の確率、突然変異したコピーを受け継ぐ50%の確率そして2つの突然変異コピーを受け継ぐ25%の確率を持ちます。この状態に苦しんで.両親が遺伝子突然変異の保因者であることが知られており、苦しむリスクが疑われる場合は、出生前診断、羊水穿刺を実施する必要があります。この技術は、酵素的および突然変異的分析のために、乳児を囲む嚢から少量の体液を除去することを含む。.診断この病状の診断は、さまざまなテストを通じて確立できます。血液、組織またはCSF(脳脊髄液)の分析は、GALC酵素の活性レベルを評価します. 非常に低いレベルまたはゼロレベルは、障害の存在を示します。このタイプの分析は診断を確認することができますが、それは病気の経過(遅いまたは速い)がどうなるかについての情報を提供しません.EEG(脳波)またはPET(陽電子放出断層撮影)のような他の試験を通して診断証拠を得ることも可能である。両方のテストはこれらの患者の異常な脳の電気的活動のパターンを示すでしょう.ニューロイメージング技術による探査もまた、障害の証拠を提供することができます。例えば、MRI / MRI(磁気共鳴画像法/機能的磁気共鳴)を通して、脳の白質の存在下で欠損を観察することができました。.疑いなくすべてのテストの中で、遺伝子の突然変異検査はこの病気の診断を確認するための最も安全で最も信頼できる技術です。さらに、その遺伝子が受けた特定の種類の突然変異に関する情報は、その障害の経過を予測するのに役立ちます。.いくつかの国では、我々が議論した検査に加えて、この病状の存在を排除するために予防検査が新生児に対して行われています。しかしながら、研究者たちはこの集団においてどの検査が最も便利であるかを見出すためにまだ努力している.クラッベ病はさまざまな時期に発症する可能性があります。影響が出生時または生後1ヶ月(1ヶ月から1年)に起こる場合、我々は早発性または乳児クラッベ病を話します. これらの子供のほとんどは、2歳になる前に死亡します。その影響が小児期(1〜8歳)の間に起こると、私たちは幼児期のクラッベ病のことを話します。最後に、この影響が8歳以降に起こると、それは若年型または成人型の遅発型と見なされ、その予後はやや致命的ではありません。.症状前述のように、この疾患(およびその他の白質ジストロフィー)は、白質またはミエリンの完全性に影響を及ぼします。ミエリンが神経系で正しい電気伝達を起こすことの重要性を知っているので、このような病気は身体に致命的な結果をもたらすと考えられます.この病状の症状は、特に病気の発症時期によって異なります。したがって、クラッベ病の出現が遅くなればなるほど、進行が遅くなり、人にとって致命的ではなくなると一般的に言われています。.クラッベ病の赤ちゃんは出生時の病気の徴候や症状がありません。実際、病気の初期段階では、医師は病理学と脳性麻痺を混同します. 最初の症状がこれらの乳児に見られ始めて、病気の異なる時期または段階で病理学の異なる絵を提示するのは、生後3〜6ヶ月までではありません。.疾患が早期発症または乳児期の場合、第一段階で症状は極度の過敏性、四肢のこわばり、頭の制御不良、断続的な親指屈曲、筋けいれんおよび高温の症状を含むことがある. 第二段階では、高張性の発作と発作、ならびに聴覚障害、視覚障害、運動障害(正しく摂食することや正しく呼吸することなど)が発生します。....

川崎病の症状、原因、治療
の 川崎病 特定のレベルでは、多系統性の血管炎であり(Delgado Rubio、2016年)、それは有機体の動脈壁の炎症を引き起こす小児の病理学である(Mayo Clinic、2014年)。.臨床的には、この疾患は動脈、リンパ節、粘膜、さらには神経系にさえも影響を及ぼします(Mayo Clinic、2014)。したがって、最も一般的な徴候や症状の一部には、通常、発熱、結膜注射、口腔異常、皮膚病理学、頸部リンパ節症などが含まれます(GarcíaRodríguezet al。、2016)。. 川崎病の起源はまだ正確にはわかっていないが、いくつかの進行中の調査はその病因を免疫学的要因および感染要因と関連づけている(Arias Cabello、FernándezÁlvarezおよびOrdaz Favila、2016).他方、早期の同定は複雑であるが、この疾患の確認は通常、臨床的および時間的経過に関する臨床基準に基づいて行われる。さらに、通常、血液分析、脳脊髄液研究または心エコー検査などのいくつかの補足的な検査があります(Bou、2014)。.治療に関しては、進行中の病状の治療と、後遺症の抑制および最小化の両方を目的とした治療的アプローチが行われています。したがって、使用される方法のいくつかは、ガンマグロブリン、アセチルサリチル酸、コルチコイドまたはモノクローナル抗体の投与を含む(Bou、2014)。.川崎病の特徴川崎病は血管炎の一種であり、主に小児人口、特に5歳未満の子供に影響を与えるまれなまたはまれな病理でもあります(The Royal Childre's Hospital Melbourne、2016).血管炎は、毛細血管、静脈または動脈の効率的な機能を変化させる、身体の血管の病理学的炎症を含む一種の障害である(National Institutes of Health、2016)。.私たちの循環器系のさまざまな構造は、血液の運搬と運搬を担当しているため、体全体に栄養分と酸素が存在し、生存に不可欠です。.したがって、感染プロセス、免疫システムの異常、またはその他の病状に関連する要因が血管内の炎症プロセスの発生をもたらす場合、これらはさまざまな方法で変更される可能性があります(National Institutes of Health、2016)。1つまたは複数の身体部位への血液の通過を妨げる、血液チャネルの部分的または完全な閉鎖.動脈瘤または奇形の発生を促進する、血液導管の壁の膨張または弱化.このように、血流が正常にすべての臓器に届かない場合、多種多様な医学的病状が起こり、それは患部によって異なる。.医学的合併症のいくつかは、とりわけ、心臓の変化、発熱、動脈瘤による採血、脳卒中などを含み得る。....

ハンチントン病の症状、原因、治療
の ハンチントン病 (EH)または ハンチントンの舞踏病, それは神経学的な遺伝性の慢性疾患です(AsociaciónCorea de HuntingtonEspañola、2016).ハンチントンは、運動障害、認知障害および精神障害の進行性の出現を特徴とする神経変性障害であり(ASHA、2016)、その人の能力および生活の質に強い影響を与える(Mayo Clinic、2014)。. この病状の症状は、運動制御、認知および行動の3つの分野に現れます。言葉の表現と表現、飲み込み、および/または運動協調の欠如における困難を観察することは可能です。さらに、記憶力の欠如、順序付け、推論、学習能力、問題解決についても説明する(ASHA、2016)。.これとは別に、ハンチントン病の最も特徴的な症状は舞踏病であり、それは非常に限定的になるまで進行する観察不能な運動不能から始まる変化です(Ramalle-Gómara、et al。、2007)。.一般に、ほとんどの場合、症状は30年から40年の間に発症し始めます(Mayo Clinic、2014)。そして、さまざまな段階の深刻さを通して段階的に発達する.症状を制御するためのさまざまな有益な薬理学的介入が記載されているが、それは致命的な疾患である(Mayo Clinic、2014).ハンチントン病(HD)の特徴の ハンチントン病 (EH神経変性、遺伝的に受け継がれた常染色体優性(Ramalle-Gomara et al。、2007)であり、そして致命的な疾患は神経学的、精神医学的および認知的変化を通して現れる(Arango-Lasprilla et al。、2003).この病状の最初の説明は、1872年にジョージ・ハンチントン博士によってなされ、そこから名前が付けられています(Arango-Lasprilla et al。、2003)。.最初の臨床参考文献は「韓国?特徴的な形で現れる不随意運動に基づいた、Huntingtonの研究(Arango-Lasprilla...
ゴーシェ病の症状、原因、治療
の ゴーシェ病 (EG)は、無症候性の形態から重度の神経学的状態までのその臨床的不均一性を特徴とする遺伝的起源の病理である(Giraldo et al。、2008).生体分子レベルでは、ゴーシェ病はベータ - グルコセレブロシド化されたリソソーム酵素の不十分な活性によって引き起こされ、その結果として、肝臓を含む様々な構造におけるグルコシルセラミド(脂肪性物質)の異常な貯蔵をもたらす。脾臓、骨、肺または神経系(Capablo Liesa et al。、2011). 一方、臨床レベルでは、徴候や症状は小児期または成人期から発生し始めることがあり、それらのいくつかは骨病変、奇形および内臓病変および/または神経損傷を含む(National Institute of Neurological Disorder and Stroke、2016)。 ).このように、この病状の診断的疑いは臨床所見、内視鏡検査の存在、骨痛、貧血などに基づいてなされ、診断は酵素活性の研究を通して確認される(Gilando、2011)。.現在、ゴーシェ病のために設計されたいくつかの治療アプローチがあり、それらのほとんどは酵素補充を目的としていた.それは冒された人々のための重要な利益を報告しますが、いくつかのケースでは、この病理学に関連した二次的な神経学的合併症はまだ現れます(Giraldo et al。、2008)。.ゴーシェ病の特徴ゴーシェ病は代謝異常であり、一般集団では稀であり、遺伝的遺伝であり、その特徴的な徴候および症状は不十分な酵素活性に起因する(National...
ファール病の症状、原因、治療
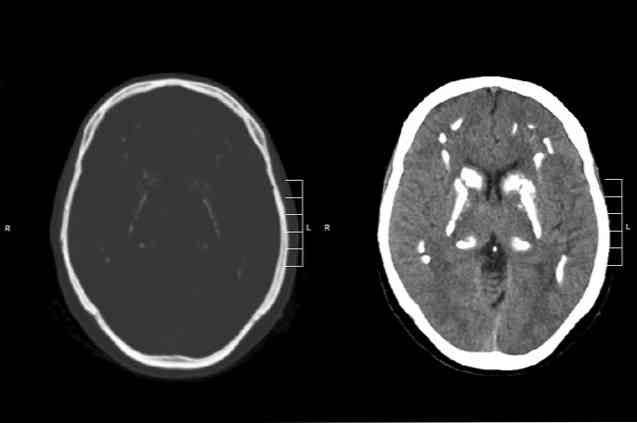
の ファール病 脳の石灰化の発生に関連する遺伝的な遺伝的起源の病理学である(Polo Verbel、Torres Zambrano、Cabarcas Barbosa、Navas、González、MontoyaおよびBolañosGarcía、2011)。.この障害は主に神経学的および精神障害の存在によって特徴付けられる。それらのうちのいくつかは、精神機能の悪化、運動の変化、または行動異常に関連しています(Polo Verbel et al。、2011)。. この病気の具体的な原因は正確にはわかっていません(Pérezet al。、2012)。 Oviedo GamboaやZegarra Santiesteban(2012)などの何人かの著者は、その病因を染色体14に位置する遺伝的異常と関連付けています。とZegarra Santiesteban、2012).この神経変性疾患の診断は、主に神経画像検査の使用に基づいています(PérezMaciá、MartínezCortés、Pecino Esquerdo、GarcíaFernández、2012)。.通常、選択された技術はコントラストのないコンピュータ断層撮影です(PérezMaciáet al。、2012)。.Fahr症候群に対する治療法も、特定の治療法もありません(Lacoma Latre、SánchezLalana、RubioBarlés、2016)。.対症療法およびリハビリテーション医学的アプローチが通常用いられる。しかしながら、ファール症候群に罹患している人々の予後は好ましくない(Oviedo Gamboa and...
ファブリー病の症状、原因、治療
の ファブリー病 は、生物のさまざまな構造における特定の種類の脂質の蓄積に関連する遺伝病理学です(Genetics Home Reference、2016)。.この病状は、X染色体に関連する遺伝的起源を有し、その臨床的特徴は、不十分なレベルのα-ガラクトシダーゼ酵素の存在によって引き起こされる(Martínez-Mechónet al。、2004)。. ファブリー病(PE)の臨床経過は、特に男性において、人生の早い時期に発生する可能性があり、神経因性疼痛、多汗症、皮膚病変、角膜異常、疲労感、疲労感などの徴候や症状を含むことがあります。聴力、心不全、腎不全および/または脳血管障害(Guelbert et al。、2015).PEの深刻な多体系的影響は、生活の質の著しい悪化を引き起こし、それは他の二次的な医学的病状の発達およびさらには罹患者の早期死亡にさえつながり得る(Barba Romero et al。、2012)。.PEの診断は通常、定義された臨床像の存在に基づいて行われ、さらに、彼らは通常、病理学を確認するために酵素活性の実験室分析および遺伝子研究を使用する。.PEで使用される治療は、二次的な医学的合併症の発症を予防し、そして酵素的欠損を補うことを目的としている(Ortiz andMarrón、2003)。この場合、酵素置換による治療的介入は、希望と生活の質を延ばすのに広く有効であることが示されている(Barba Romero et al。、2012)。.ファブリー病の特徴ファブリー病(EF)は遺伝病理学であり、その症状はさまざまな臓器の細胞内の脂質沈着物および私たちの体の構造の蓄積の結果として起こります(Genetics Alliance、2016).体内には何千もの活性物質があり、その中に酵素があります。酵素は、ある種の生化学反応の調節および/または促進において重要な役割を担っているタンパク質分子の一種です。. したがって、例えば、腸の構造の中には、消化過程や生物の基本的な栄養素の抽出を制御するために、私たちが食物を分解するのを助ける酵素があります(Genetics Alliance、2016).さらに、これらの酵素の大部分は、リソソームと呼ばれる細胞構造の特定の領域に保存されています。これは、脂質、炭水化物およびタンパク質の分解に対するサポートを身体に提供することができる(Genetics Alliance、2016).このように、ファブリー病において、脂質および他の類似物質の代謝に不可欠な、α-ガラクトシダーゼと呼ばれる酵素の機能または欠如は不十分な分解を引き起こす。.したがって、脂質は、神経系、心血管系、無感覚性、眼球性などのような体のさまざまな領域に蓄積する傾向があります。...

シャルコー・マリー・トゥース病症状、原因、治療
の シャルコーマリートゥース病 それは、感覚運動性多発ニューロパチー、すなわち末梢神経の損傷または変性を引き起こす病理学である(National Institutes of Health、2014)。それは遺伝的起源の最も頻繁な神経学的病状の1つです(国立神経疾患研究所および脳卒中、2016年).1886年に初めて説明した3人の医師、Jean-Martin Charcot、Pierre de Marie、およびHoward Henry Thoothからその名前が付けられました(Muscular Dystrophy Association、2010)。. それは、感覚および運動症状が現れる臨床経過を特徴とし、それらのいくつかは上肢および下肢、特に足の変形または筋力低下を含む(Cleveland Clinic、2016)。.さらに、末梢神経の機能と構造に関連するタンパク質の産生を担う遺伝子にさまざまな突然変異が存在することによって引き起こされる遺伝病です(国立神経障害脳卒中研究所)。, 2016).一般的に、シャルコー - マリー - トゥース病の特徴的な症状は青年期または成人期の初期に現れ始め、その進行は通常緩やかである(国立神経疾患研究所および脳卒中、2016)。.この病状は一般に罹患者の生命を危険にさらすことはありませんが(Muscular Dystrophy...

カナバン病症状、原因、治療
の カナバン病 それは脳内の神経細胞が損傷を受けていて互いに連絡できないために起こるまれな遺伝病です.アシュケナージユダヤ人集団(中央ヨーロッパの東部に定住)およびその子孫でより頻繁に発生しているが、この疾患はあらゆる社会および民族のグループに存在し、6,400-13.00人に1人が罹患している。世界的な罹患率は不明です. カナバン病の特徴この病気は白質ジストロフィーのグループ内にあります。この範疇は、ニューロンの軸索を囲むミエリン鞘が損傷を受けており、したがってニューロン間の良好なコミュニケーションがない全ての遺伝的障害を包含する。.この病気の最も一般的な、そして同時に、最も深刻な形態は新生児または乳児です。カナバン病のこの形態は、生まれたばかりの子供たちまたは彼らの人生の最初の数年間に影響を及ぼします.この病気にかかっている子供たちは人生の最初の数ヶ月の間に何の問題もありませんが、これらは3から5ヶ月の間に咲き始めます.主な症状は発達障害が原因で発生します。子供たちは運動障害を抱えて、転倒したり、頭を回転させたり、何の支えもなく座ったりできなくなります.他の一般的な症状は、筋力低下(筋緊張低下)、頭部の異常発達(大頭症)および過敏性です。それほどではありませんが、食事、発作、睡眠障害などの問題もあります。.それほど一般的ではないもう1つの形態は、カナバンの小児中年期または青年期発症の疾患です。この疾患を持つ子供や青年は言語発達や運動能力に問題がありますが、これらの問題はしばしば非常に軽度なのでカナバン病の症状として識別されません。.カナバン病を患っている人の平均余命は非常に不均一であり、発症の時期によって大きく異なります.新生児または乳児の形態に苦しむ子供たちは、通常、ほんの数年で暮らしますが、中には思春期に達し、成人するまではごくわずかです。少年型に苦しむ人々は通常の平均余命を持っていますが.症状既に述べたように、カナバン病には2つの高分化型があります:新生児期または小児期の発症と中年期または青年期の発症. 新生児または乳児の開始カナバン病の新生児期または乳児期発症の症状は非常に深刻で、通常3〜50ヶ月齢まで気付かれず、大頭症、頭の運動制御の喪失、および発達障害を含む。子供が成長するにつれて発達障害がより明白になる.最も深刻な症状は運動障害に関連したものです、なぜなら子供たちはサポートなしでは座ることも立ち上がることもできず、歩くことも話すこともできないからです。彼らが年配の低緊張になると痙縮が起こります.たとえ彼らがこれらのすべての運動問題を抱えていても、彼らは社会的に相互作用することを学ぶことができます、微笑み、対象を指摘してください... 彼らはまだ視覚的にオブジェクトを識別することができますが、いくつかの子供たちはまた、視覚的な問題を引き起こす視神経萎縮症に苦しんでいる.症状が悪化すると、症状は悪化し、睡眠障害、発作、摂食障害を引き起こします。子供は完全に頼りになり、どんな仕事をするのにも助けが必要です.これらの子供たちの平均余命はかなり短く、ほとんどは数年で死亡しますが、中には青年期または成人期まで生きる人もいます.平均的な小児期または青年期カナバンの小児中年期または青年期発症の疾患は、以前のものより軽度です。症状には、言語および運動発達におけるいくつかの困難が含まれます.カナバン病の症状として識別されないほど通常それらは穏やかですが、マーカーの一つは高濃度のN-アセチルアスパラギン酸(NAA)であるため、この疾患は通常尿検査を行った後に診断されます。 、英語でのその頭字語のために)尿中.原因この病気はASPAと呼ばれる遺伝子の突然変異によって引き起こされます。この遺伝子はNAAの分子を分解するために責任がある酵素のアスパルトアシラーゼの制御である.ASPA遺伝子の突然変異はアスパルトアシラーゼの有効性を低下させるため、十分なNAA分子を分解することはなく、この物質が高濃度になります。この突然変異が早く起こるほど、それが持つ悪影響はひどくなります.NAA分子の機能はよく理解されていないが、それらはニューロンを通る水分子の輸送に関与しているようであり、そしてこの物質の過剰は新しいミエリンの形成を妨げそして現存するものを破壊する。これは、ニューロン間の接続が正しく機能せず、脳が正常に発達できないことを引き起こします。.さらに、この疾患は常染色体劣性の方法で遺伝する可能性があります。したがって、カップルの各メンバーがASPA遺伝子の病原性多様体の保因者であり、子供をもうけることにした場合、彼らは以下のようなことをする可能性があります。小児は25%の症例でこの疾患を示している.子供は50%のケースで保因者ですが、問題はありません.息子は25%もキャリアではありません. 危険にさらされている人口、この場合はアシュケナージユダヤ人の子孫である個体が、彼らが子供を持つ前にASPA遺伝子の保因者であるかどうかを調べるための遺伝分析を持つことは非常に重要です.治療治療法は、病気の形態や各個人が示す症状によって異なります. 新生児または乳児のカナバン病の治療現在、カナバン病の治療法はありません。そのため、利用可能な治療法は、サポート、栄養補給、水分補給、感染症の予防と治療による患者の生活の質の向上に焦点を当てています.姿勢や運動能力を向上させ、拘縮や褥瘡などの筋肉の問題を回避し治療するために、子供たちは理学療法を受けることをお勧めします。コミュニケーションスキルを向上させるための治療プログラムや教育プログラムに参加することもできます。.小児が発作を起こしている場合は、薬物による治療に抗てんかん薬(AED)、アセタゾラミド(商品名Diamox)が含まれます。®)頭蓋内圧とボツリヌス毒素(ボトックス)の注射を減らすために®)痙縮がある場合はそれを治療する.それは子供の状態と彼の発達がどのように進んでいるかをチェックするために6ヶ月ごとにフォローアップする必要があります.中年期または青年期のカナバン病の治療この種の病気に苦しむ人々ははるかに穏やかな症状を経験するので、彼らは通常彼らの言語または特別な教育プログラムを改善するために治療を必要とするだけです。彼らは薬を必要としません. 子供の状態を年1回モニタリングすることをお勧めします.新しい治療法ヒトと動物モデルの両方における他の治療法の有効性が現在研究されています.人間との研究- 非ウイルスベクター非ウイルスベクターを使用して、カナバン病の子供の脳で遺伝子移植の有効性が調査されています.最初の結果は、この種の移植は小児によく耐えられ、そしていくらかの生化学的、放射線学的および代謝的変化を引き起こすことを示しているが、それでも疾患を治癒するのには有用ではない。 2002年まで).- ベクトルVAAV2McPhee等。 (2006)健康なASPA遺伝子がベクトルとしてAAV2を使って、子供の体のいくつかの場所に移植されるという研究を行っています。 10人のボランティアの子供たちが参加したテストの1つで。そのうちの3人において、移植はその抗体を働いて中和しました、しかし、子供たちのどれも改善しませんでした.- クエン酸リチウムクエン酸リチウムは、脳内のNAA濃度のレベルを下げることができます。 (2010)彼らが60日間カナバン病を持つ6人にクエン酸リチウムを投与する実験を実施することにしました.臨床的改善は見られなかったが、大脳基底核および前頭葉の白質中のNAAの濃度レベルが判明した。.- グリセロールトリアセテートアスパルトアシラーゼ酵素の欠如は脳内の低レベルのアセテートを引き起こすので、Mahavaraoと彼のチーム(2009)はアセテートレベルを上げるためにグリセロールトリアセテートを2人のカナバル病患者に投与し、それが増加するかどうか確認アスパルトアシラーゼのレベルも.臨床上の改善は見られなかったが、化合物は患者によって十分に許容された。彼らは現在、より多くの量のグリセロールトリアセテートを投与することによってテストを行っています.動物を使った研究病気を表す動物モデルを作成する方法の1つは動物を作成することです ノックアウト. これらの動物、通常はマウスは、疾患で変化している遺伝子を除去または変更するように遺伝子操作されています。この場合、修飾遺伝子はASPA遺伝子です。.動物モデルは、疾患をよりよく理解し、その生物学的相関を研究し、そして新しい治療法の有効性をチェックするのに役立つ。.マタロン等。...
